idiopathic inflammatory myositis
- related: Rheumatology
- tags: #literature #pulmonology
The patient has an idiopathic inflammatory myositis (dermatomyositis, polymyositis, or antisynthetase syndrome), with the myositis panel demonstrating the presence of a PL-7 antisynthetase antibody and a 52kD SS-A (Ro) antibody. The diagnosis was suggested by the fever nonresponsive to antibiotics; rash (although antibiotic drug reaction could also explain this finding); proximal muscle weakness; difficulty swallowing; elevated C-reactive protein level and abnormal liver enzymes, which can also be elevated in myositis; the elevated aldolase level, which does not always track with the creatine kinase level; and unexplained patchy ground-glass opacities in a pattern suggestive of organizing pneumonia (Figure 3). The presence of the myositis-specific antibody PL-7 confirms the diagnosis, with the myositis-associated 52kD SS-A (Ro) antibody often present in these patients as well (Figure 4). Antinuclear antibody testing should be performed, but the result is often negative in patients with an idiopathic inflammatory myositis because the autoantigens are expressed in the cell cytoplasm and, if positive, would not delineate the precise rheumatologic diagnosis. Although the water in the basement suggests that a search for mold inhalation may be warranted, the clinical presentation is not that of hypersensitivity pneumonitis, and the hypersensitivity antibody panel is neither sensitive nor specific in making that diagnosis. Immunoglobulin levels are not indicated in this setting.
Failure to make the diagnosis and institute therapy can result in rapid progression to acute hypoxemic respiratory failure. Pulmonary fibrosis with a nonspecific interstitial pneumonia pattern may develop in areas of previous organizing pneumonia; this overlap is a radiographic hallmark of interstitial lung disease associated with an idiopathic inflammatory myositis. Although systemic corticosteroids often result in improvement, most patients require multimodality immunosuppressive therapy (Figure 5).
The patient improved with corticosteroids and the addition of mycophenolate mofetil. His dyspnea markedly decreased, exercise ability increased, and dysphagia and weakness resolved, and he no longer required home oxygen (Spo2 breathing room air was 95% at rest and 92% after walking for 6 min). When he was last assessed, his FVC had increased to 75% predicted from 62% predicted. His chest CT scan showed improvement in the ground-glass opacities (Figure 6). Unlike classical dermatomyositis, which in older adults may be paraneoplastic, interstitial lung disease related to the antisynthetase syndrome is less often associated with malignancy. In this patient, it is tempting to speculate that his presentation is a paraneoplastic process because the initial phase of the interstitial lung disease was present on the chest CT scan obtained before colon resection (Figure 7). Screening results for an occult second malignancy were negative.
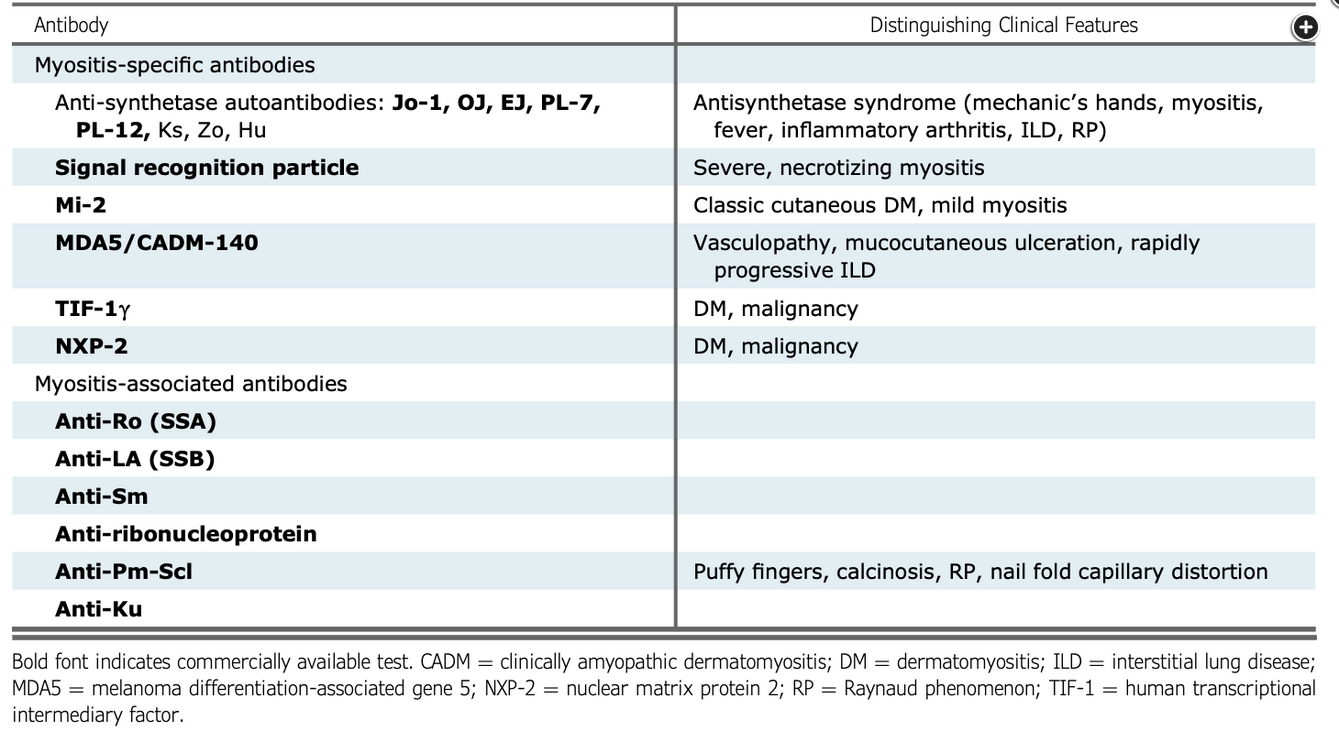
Myositis-specific and associated antibodies.
Figure courtesy of Jablonski R, Bhorade S, Strek ME, et al. Recognition and management of myositis-associated rapidly progressive interstitial lung disease. Chest. 2020;158(1):252-263.
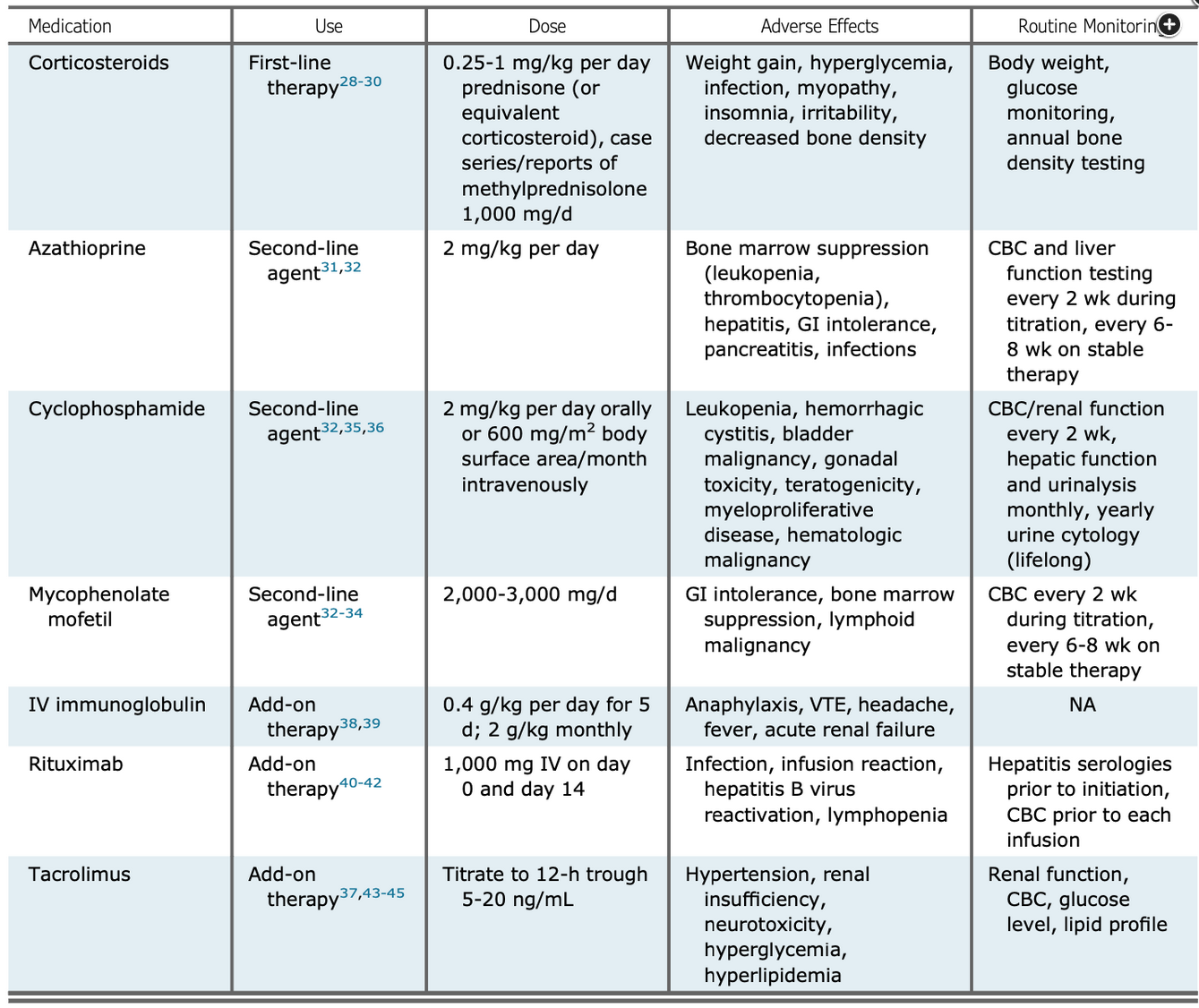
Treatments used for idiopathic inflammatory myositis.
Figure courtesy of Jablonski R, Bhorade S, Strek ME, et al. Recognition and management of myositis-associated rapidly progressive interstitial lung disease. Chest. 2020;158(1):252-263.