pulmonary alveolar proteinosis
- related: ILD Interstitial Lung Disease or Diffuse Parenchymal Lung Disease
- tags: #literature #pulmonology
- disease: 1.pulmonary alveolar proteinosis
- antibody: 2.GM-CSF
- BAL finding: 2.classy milky white appearance
- pathogenesis: clearance of surfactant altered
- etiology: congenital, autoimmune, exposure
- patient: middle age, dyspnea, productive cough
- CT: crazy paving
- BAL: classy milky white appearance, positive PAS stain extracellular material, intraalveolar lipoproteinaceous material
- GM-CSF antibodies (both sensitive and specific)
- treatment: sequential lavage (old), inhaled GM-CSF, rituximab, molgramostim
Source
The patient has pulmonary alveolar proteinosis (PAP) on the basis of a typical presentation in middle age of dyspnea and productive cough associated with a chest CT scan showing ground-glass opacities with interlobular and intralobular septal thickening, which characterizes the “crazy-paving” sign (Figure 2). Patients with PAP will exhibit circulating antibodies to granulocyte-macrophage colony-stimulating factor (GM-CSF) (choice A is correct).
While chest CT findings in PAP can be highly suggestive of the diagnosis, they are nonspecific and can be seen with inhalation of dusts, including silica; Pneumocystis pneumonia; ARDS; cardiogenic pulmonary edema; lipoid pneumonia; medication pneumotoxicity; and acute exacerbation of an underlying interstitial lung disease. The diagnostic algorithm for PAP recommends first performing bronchoscopy with BAL, which is inspected for the classic milky white appearance and is sent for periodic acid-Schiff (PAS) stain, which is positive in PAP (Figure 3). Transbronchial biopsy is recommended if this can be done safely, to look for intraalveolar deposits of lipoproteinaceous material (Figure 4). GM-CSF antibody is present in the serum in the majority of patients with adult-onset PAP. GM-CSF antibodies can also be found in the BAL fluid and signifies the PAP is autoimmune in etiology. A positive serum GM-CSF antibody along with compatible high-resolution CT imaging and BAL fluid with PAS-positive proteinaceous material allows a confident diagnosis of autoimmune PAP. While surgical lung biopsy is recommended in cases in which less invasive evaluation has been unrevealing, it is necessary in only a minority of patients and can be nondiagnostic if the lung disease is patchy, as was the case in this patient.
PAP is a disorder of surfactant homeostasis in which production and/or clearance of surfactant is impaired. This can be a congenital process due to genetic abnormalities that result in decreased surfactant production; autoimmune in nature from antibody production to GM-CSF; or secondary to inhalational exposures to dusts, especially in the workplace or as a result of hematologic malignancy. In adults with PAP, >90% of the cases are autoimmune and characterized by the presence of a serum GM-CSF antibody, which prevents the differentiation of macrophages, which are essential for the clearance of surfactant protein degradation products and require GM-CSF to perform this function. This leads to the accumulation of lipoproteinaceous material in the alveoli, which causes the typical clinical and radiographic findings. Treatment depends on the type of PAP that is present, with whole lung lavage the standard therapy, which was performed sequentially (right then left lung) in this patient and was curative. Because the autoimmune defect typically leads to recurrent lung disease and eventually chronic hypoxia, pulmonary fibrosis, and pulmonary hypertension with right heart dysfunction, a search for medical therapy has led to the development of supplemental GM-CSF therapy, which has been shown to be clinically effective when given subcutaneously, and by inhalation of molgramostim. Rituximab has also been used to decrease autoantibody formation.
For decades, the mainstay of treatment for PAP has been whole lung lavage, which entails periodic intubation of the patient with a double-lumen tube, isolation of each lung, and large-volume lavage of a single lung during each procedure to clear accumulated surfactant. Once the molecular basis and the role of GM-CSF was elucidated, therapies to augment reduced GM-CSF action have been studied, initially in small series and then in randomized trials. In the IMPALA trial, placebo, continuous (daily), and intermittent (daily for a week every other week) GM-CSF augmentation were compared. Patients received placebo or drug by a simple nebulization device that could be used at home or in the outpatient clinic. Treatments were given for 24 weeks, and then the active drug treatments extended for an open-label period. Daily inhaled but not intermittent use resulted in improvement in diffusing capacity, CT ground-glass opacities score, and St. George’s Respiratory Questionnaire dyspnea score. Inhaled molgramostim for GM-CSF augmentation is available for off-label use at the time of this publication. Another trial extending the treatment period of inhaled molgramostim is underway.

BAL cytology shows scanty macrophages in abundant periodic acid-Schiff–positive extracellular material.
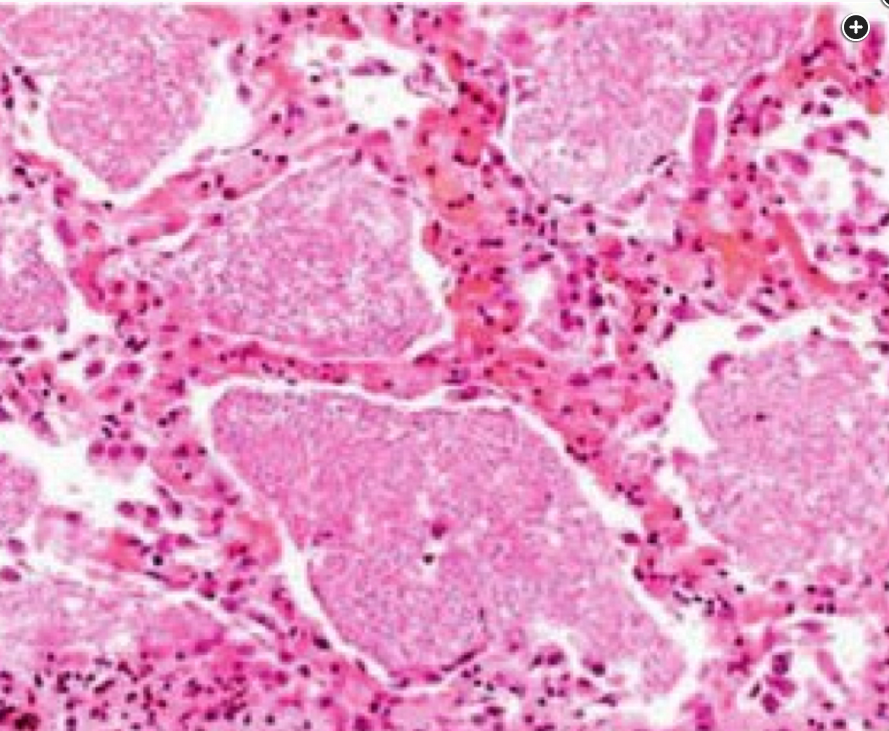
Histopathology (hematoxylin and eosin stain, ×100 magnification) shows intraalveolar deposits of lipoproteinaceous material circumscribed by thickened alveolar walls owing to chronic inflammatory cell and type 2 pneumocyte hyperplasia lining the walls.
Links to this note
-
ILD Interstitial Lung Disease or Diffuse Parenchymal Lung Disease
- crazy paving pattern: pulmonary alveolar proteinosis
-
alveolar proteinosis have crazy paving patterns
- related: pulmonary alveolar proteinosis