GBS
- related: Neurology, ICU intensive care unit
- tags: #neuro #literature #pulmonology
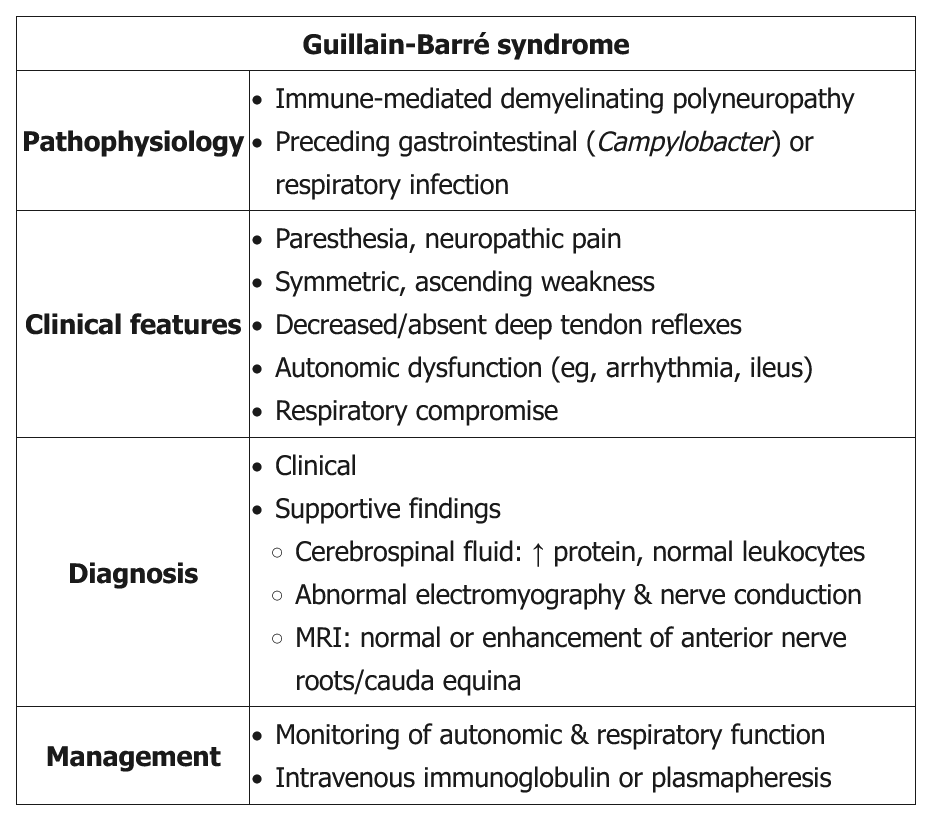
Classic GBS (AKA AIDP) presents as a progressive, symmetric ascending weakness or paralysis, often a few days to a week after a respiratory or gastrointestinal infection (especially Campylobacter jejuni). About one-third of patients have no history of antecedent illness. As the weakness ascends (eg, difficulty walking with 3/5 strength in the lower extremities compared with 4/5 strength in the upper extremities in this patient), bulbar symptoms (eg, ophthalmoplegia, facial diplegia, dysarthria, dysphasia) and respiratory compromise can develop. Back pain can be a presenting feature of GBS as the disease targets primarily the proximal nerves and nerve roots in either the cervical or lumbar spine. Although GBS is primarily a motor polyneuropathy, patients can have mild sensory symptoms such as paresthesias and sensory ataxia. GBS patients also are at risk of developing dysautonomia (eg, tachycardia, diaphoresis, sluggish pupils). Examination shows absent reflexes.
Lumbar puncture is indicated in all suspected GBS patients to rule out infectious etiologies and document elevated cerebrospinal protein with a normal cerebrospinal fluid white blood cell count (albuminocytologic dissociation). However, lumbar puncture may not be diagnostic in some GBS patients as it may take up to a week after symptom onset to see the abnormal findings. Nerve conduction study and electromyography are also indicated to confirm the diagnosis and estimate prognosis.
Treatment is mainly supportive, with plasma exchange or intravenous immunoglobulin. Current evidence does not recommend corticosteroids as they are not beneficial. Most patients recover within several weeks to months.
- high risk of respiratory failure: frequent measure vital capacity and negative inspiratory force (same for myasthenia crisis)
- plasma exchange or IVIG to combat antibodies against nerves
- Patients with GBS should receive plasma exchange or IVIG if:
- Nonambulatory
- Within 4 weeks of symptom onset
- Those who are ambulatory and recovering generally do not require treatment.
- Transverse myelitis is typically treated with high-dose glucocorticoids, not GBS
Prognosis
The manifestations of Guillain-Barré syndrome (GBS) tend to evolve as follows:
- 2 weeks of progressive motor weakness that can lead to paralysis
- 2-4 weeks of plateaued symptoms
- Slow, spontaneous recovery over months
At a year after symptom onset, 85% of patients with GBS have regained the ability to walk and nearly 60% have had full, spontaneous neurologic recovery. Although patients with GBS typically recover without intervention, treatment with plasma exchange or intravenous immunoglobulin shortens the time to recovery by approximately 50%.
- LP: elevated protein, normal WBC
Autonomic dysfunction as well as back pain and spasm may precede weakness in 30% of individuals experiencing Guillain-Barre Syndrome (GBS), a rare condition estimated at 1 to 2 cases per 100,000. Of the available options, plasmapheresis would be considered the treatment of choice for this condition (choice A is correct). Although plasma exchange may not be ideal for individuals who are hemodynamically unstable, in this case, the patient had been stabilized.
GBS can affect all age groups but more commonly affects men. The condition is thought to result from an immune response to a preceding infection that cross-reacts with peripheral nerve components because of molecular mimicry. The immune response can be directed toward the myelin or the axon of peripheral nerve, resulting in demyelinating and axonal forms of GBS. Campylobacter jejuni infection is the most commonly identified infectious agent that can precipitate GBS, with cytomegalovirus, Epstein-Barr virus, human immunodeficiency virus, and Zika virus having also been associated with the condition. Some patients develop GBS after another triggering event such as immunization, surgery, trauma, medications, or bone-marrow transplantation.
Typically, symptoms progress over a few days to a week. The presence of symmetric progressive muscle weakness with loss of patellar reflexes is suggestive of the condition. Autonomic dysfunction is a well-recognized feature of GBS and is a significant source of mortality. Dysautonomia occurs in 70% of patients and manifests as symptoms that include tachycardia (the most common) and bradycardia, urinary retention, hypertension alternating with hypotension, ileus, and loss of sweating. Back pain due to nerve root inflammation can be a presenting feature and is reported during the acute phase in two-thirds of patients with GBS. Ophthalmoplegia with ataxia and areflexia may be seen in Miller-Fisher Syndrome (MFS), considered to be a more limited variant of GBS. In 25% of patients who present with MFS, some extremity weakness is noted. Of note, some patients with MFS develop fixed and dilated pupils, and antibodies against GQ1b (a ganglioside component of nerve) are present in 85 to 90% of patients with each syndrome.
Use of intravenous immunoglobulin administration could also be considered. Patients with GBS treated early with this or plasmapheresis can often experience rapid resolution of their symptoms, and it is estimated that the time to full recovery can be shortened by 40%. In this case, although the pulse rates were sporadic and dipped below 30/min, there is no indication to utilize atropine in this situation as the vital signs have already been stabilized (choice B is incorrect). Back pain due to nerve root inflammation can be a presenting feature and is reported during the acute phase in two-thirds of patients with GBS. Although corticosteroids have anti-inflammatory properties, treatment with glucocorticoids in adults with GBS is not recommended (choice C is incorrect). Thiamine, formerly known as vitamin B1, is involved in the conversion of pyruvate to acetyl CoA as well as many other cellular metabolic activities, and lack of thiamine can result in peripheral neuropathies. Although TPN was being administered in this case, there is no evidence for deficiency in this vitamin and supplementing thiamine in light of the other findings would not be the best option (choice D is incorrect).1
Links to this note
-
- related: Neurology, GBS, myasthenia gravis
-
- Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is a potentially treatable autoimmune neuropathy. CIDP often presents with a progressive or relapsing symmetric proximal and distal weakness and sensory symptoms with diffuse areflexia. Its temporal course is often subacute with progression after 8 weeks of onset, which helps to differentiate CIDP from acute inflammatory demyelinating polyneuropathy (AIDP aka GBS). Atypical forms include multifocal asymmetric and distal symmetric variants, but respiratory failure is rare. CIDP may be isolated or occur in the setting of several other systemic conditions, including diabetes mellitus, lymphoma, and HIV. CSF findings are similar to those of GBS and may show albuminocytologic dissociation, and a demyelinating pattern on EMG is the key to diagnosis. Nerve biopsy is often unnecessary but, in complex presentations, can differentiate CIDP from vasculitis or amyloidosis. First-line treatments include glucocorticoids, periodic IVIG, or plasma exchange, which all have similar efficacy. Treatment is typically continued for at least 6 months. Half of patients achieve remission, but the other half relapse and require resumption of immunotherapy. Second-line therapies, including azathioprine, mycophenolate mofetil, cyclosporine, or cyclophosphamide, are often used off-label to treat refractory disease.